Tecnologia Científica
Uma maneira melhor de modelar o comportamento de ligas metálicas
A abordagem dos pesquisadores do MIT captura padrões atômicos sutis, melhorando as previsões das propriedades dos materiais.
Por
Zach Winn - 25/06/2026


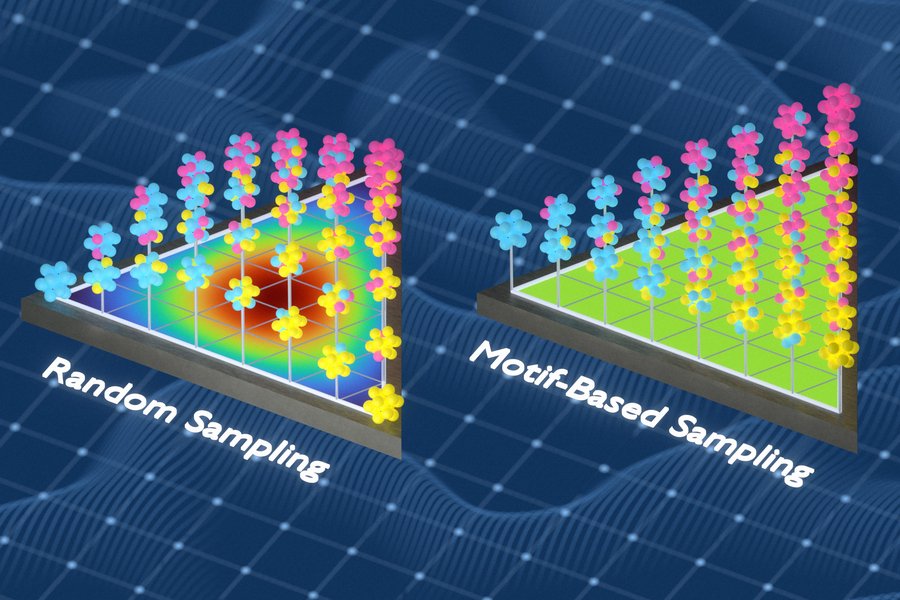
Pesquisadores do MIT criaram uma técnica que captura arranjos químicos em materiais para melhorar as previsões de como ligas metálicas e outros materiais complexos irão se comportar. Esta figura compara uma abordagem de amostragem aleatória com a nova amostragem baseada em motivos desenvolvida pelos pesquisadores. Crédito: Cortesia dos pesquisadores
Empresas que atuam na vanguarda dos setores aeroespacial, energético e da computação estão constantemente em busca de novos materiais para melhorar o desempenho. Mas, para entender como esses materiais se comportarão na prática, dentro de foguetes ou em chips de computador, as empresas primeiro precisam produzi-los e depois testá-los. Isso porque mesmo as técnicas de simulação mais poderosas têm dificuldade em modelar os complexos arranjos químicos da maioria dos materiais sólidos atuais. Esse problema aumenta os custos e o tempo da inovação em materiais.
Agora, uma equipe de pesquisadores do MIT criou uma maneira de modelar com precisão o comportamento de metais, independentemente da complexidade de sua estrutura química. No centro dessa abordagem estão modelos de aprendizado de máquina que tornam as simulações de materiais mais rápidas e precisas. Os pesquisadores aprimoraram esses modelos criando conjuntos de dados de treinamento que capturam a diversidade de ambientes atômicos em materiais quimicamente desordenados.
Em um novo artigo publicado na revista Science Advances , os pesquisadores demonstraram que sua abordagem pode ser usada para prever com precisão as propriedades de materiais para um grupo diversificado de ligas metálicas sob uma variedade de condições. Eles também mostraram como a abordagem pode ser usada para desenvolver novos materiais, especialmente em cenários onde a experimentação é dispendiosa.
“O foco do artigo são as ligas metálicas, que é a área em que trabalho, mas isso poderia ser adaptado a outros tipos de materiais, como semicondutores”, diz o autor sênior Rodrigo Freitas, professor de Desenvolvimento de Carreira TDK em Ciência e Engenharia de Materiais do MIT. “Não se limita a uma única aplicação — você poderia usar essa abordagem para criar novos aços sustentáveis, novos materiais para a indústria aeroespacial e muito mais. É isso que torna tudo tão empolgante.”
Além de Freitas, o artigo conta com a colaboração do primeiro autor Killian Sheriff, PhD '26; dos estudantes de doutorado do MIT Daniel Xiao e Yifan Cao; e do professor sênior da Universidade de Sheffield, Lewis R. Owen.
Modelagem de metais
As propriedades dos materiais são determinadas principalmente pela organização interna de seus elementos químicos. Mesmo que dois materiais tenham a mesma composição química, diferentes arranjos químicos podem fazer a diferença entre um material quebradiço e um que se deforma sem quebrar.
Capturar essa distinção exige a simulação de materiais átomo por átomo. Para isso, os pesquisadores se baseiam em modelos que descrevem como os átomos interagem entre si. Nas últimas duas décadas, o aprendizado de máquina se tornou a maneira mais precisa de construir esses modelos. Tais modelos funcionam bem quando os arranjos químicos dentro dos materiais seguem padrões altamente ordenados, mas esse não é o caso da maioria dos materiais sólidos, cujos arranjos químicos atômicos são desordenados e variam de uma região para outra.
“O verdadeiro desafio em nossa área é modelar essas fases quimicamente desordenadas”, diz Freitas. “A desordem química significa que existe uma enorme variedade de ambientes químicos locais, o que é difícil para o modelo de aprendizado de máquina aprender. Isso é um problema porque todos os metais que usamos na prática são quimicamente desordenados.”
O problema reside na falta de dados de treinamento representativos para essas simulações átomo por átomo. A principal abordagem atual para a criação desses dados funciona por força bruta, muitas vezes exigindo mais de 100.000 horas de computação para gerar os dados de treinamento para um único material. Mesmo assim, o resultado não é satisfatório quando os pesquisadores alteram a composição do material.
Em trabalhos anteriores , o grupo de Freitas desenvolveu um método para medir a complexidade química de materiais sólidos analisando a frequência e o espaçamento de pequenos grupos de átomos. Para este estudo, os pesquisadores utilizaram essa capacidade para construir conjuntos de dados de treinamento mais robustos. Eles empregaram uma abordagem matemática conhecida como teoria da informação para gerar conjuntos de dados de treinamento que capturam uma variedade maior de ambientes químicos locais dentro de materiais desordenados. O método funciona substituindo átomos das amostras para reduzir a repetição e expor o modelo a ambientes químicos que ele poderia, de outra forma, não detectar.
“Continuamos otimizando o conjunto de treinamento para que ele capturasse o máximo possível de ambientes locais diferentes”, diz Freitas. “Se o mesmo tipo de ambiente aparecesse muitas vezes, substituíamos os exemplos redundantes por outros que o modelo não tivesse visto antes. Isso torna o conjunto de treinamento muito mais informativo, porque cada exemplo adiciona algo novo.”
Quando treinados com os conjuntos de dados dos pesquisadores, os modelos previram as propriedades dos materiais com mais precisão do que os modelos treinados usando amostragem aleatória ou outro método de amostragem popular.
“O ponto de partida para todas essas simulações átomo por átomo é: você consegue descrever com precisão a ligação química entre os átomos?”, explica Freitas. “Caso contrário, ainda é possível aprender sobre materiais em geral, mas não se sabe o que acontecerá com materiais específicos no mundo real. Essa abordagem torna as simulações altamente fiéis em termos de química, para refletir melhor o que acontece com os materiais.”
Os pesquisadores aplicaram sua técnica para criar conjuntos de dados de treinamento de aprendizado de máquina para um grupo de ligas metálicas quimicamente diversas. Usando um conjunto de modelos de aprendizado de máquina, eles mostraram que os modelos treinados em seus conjuntos de dados são mais precisos do que modelos muito maiores criados por empresas como Google e Microsoft.
“Chegamos a um ponto em que estávamos convencidos de que funcionava sem usar esses métodos caros de força bruta”, diz Freitas. “Eu disse a Killian: 'Este é um bom artigo. Mas se você puder mostrar que as simulações com esses modelos agora podem prever com precisão propriedades úteis dos materiais, então ele se torna um artigo excelente.' Killian levou isso a sério e testou o método o máximo que pôde.”
Sheriff trabalhou com Xiao e Cao para testar a abordagem em diferentes ligas e propriedades. A equipe também utilizou os dados experimentais de Owen para comparar as simulações com medições reais da ordenação atômica em ligas.
Do laboratório à indústria
O método funciona, em parte, capturando padrões ocultos nos dados da amostra. Os pesquisadores descrevem os padrões no artigo como "tendências energéticas sutis em relação a certas configurações químicas locais".
Essas pequenas diferenças energéticas são importantes porque determinam quais fases se formam em uma liga, como essas fases mudam com a temperatura e a composição e, em última análise, quais propriedades o material terá. Como um teste, Daniel Xiao liderou simulações que mostraram que os modelos da equipe conseguiam prever diagramas de fase que correspondiam de perto aos dados experimentais. Os diagramas de fase mapeiam quais fases são estáveis ??em diferentes temperaturas e composições químicas, e são uma ferramenta fundamental para o projeto e processamento de ligas.
“Os diagramas de fases são uma das principais maneiras pelas quais as pessoas conectam a modelagem de materiais às decisões reais de processamento”, diz Freitas. “Se você estiver soldando, fundindo ou realizando tratamento térmico em uma liga, precisa saber quais fases provavelmente se formarão sob diferentes condições. Nosso objetivo é tornar esses tipos de previsões suficientemente precisos e acessíveis para que se tornem parte da maneira como as pessoas projetam materiais.”
Os pesquisadores agora estão usando essa abordagem para estudar como a alteração da composição de uma liga afeta as propriedades mecânicas e a tolerância à radiação, com o objetivo de projetar materiais que permaneçam resistentes e tolerantes a danos em ambientes hostis. Eles também estão trabalhando para tornar o método mais fácil de usar com os tipos de ferramentas e fluxos de trabalho que os engenheiros de materiais já utilizam.
“A indústria não vai mudar a forma como faz as coisas se o que você está criando não se encaixar nos procedimentos operacionais existentes”, diz Freitas. “O objetivo é tornar essas previsões úteis nos locais onde as decisões sobre materiais são de fato tomadas.”
A pesquisa foi financiada pelo Escritório de Pesquisa Científica da Força Aérea dos EUA.